★特殊染色體疾病★
(1) 普瑞得─威立氏症候群
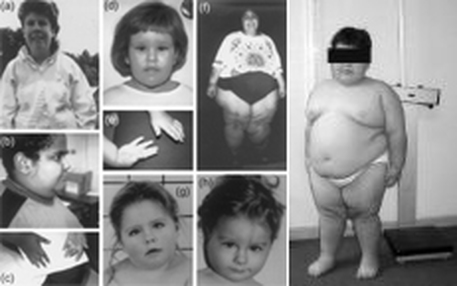
普瑞得─威立氏症候群 (Prader-Willi syndrome; PWS) 是一種特殊的染色體疾病,約有70%的患兒其來自父親的第15號染色體長臂15q11-q13位置上會有微小的脫失(microdeletion)。
其餘找不到染色體缺損的患者則被證實其兩個外觀正常的15號染色體皆遺傳自母親,而缺少了來自父親的15號染色體,此稱為單親性雙染色體症(uniparental disomy; UPD)──這個奇特的現象最可能導因於三染色體自救(trisomy rescue),或是母親卵細胞減數分裂時發生15號染色體不分離,並與缺少15號染色體的精細胞結合所致。基因組銘記作用(genomic imprinting)使得15號染色體為母源性UPD的患者,因缺乏父系基因組的調和性影響,而導致普瑞得─威立氏症候群;如果情況相反,當來自母親的15號染色體有15q11-q13的微小脫失,或15號染色體為父源性UPD,則會產生Angelman 氏症候群。
其餘找不到染色體缺損的患者則被證實其兩個外觀正常的15號染色體皆遺傳自母親,而缺少了來自父親的15號染色體,此稱為單親性雙染色體症(uniparental disomy; UPD)──這個奇特的現象最可能導因於三染色體自救(trisomy rescue),或是母親卵細胞減數分裂時發生15號染色體不分離,並與缺少15號染色體的精細胞結合所致。基因組銘記作用(genomic imprinting)使得15號染色體為母源性UPD的患者,因缺乏父系基因組的調和性影響,而導致普瑞得─威立氏症候群;如果情況相反,當來自母親的15號染色體有15q11-q13的微小脫失,或15號染色體為父源性UPD,則會產生Angelman 氏症候群。
★常見的症狀有哪些?
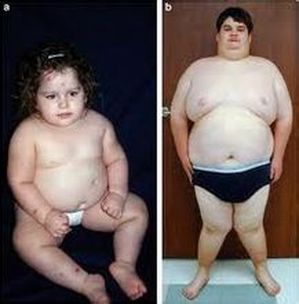
PWS患兒的肌肉張力差、皮膚及頭髮顏色較淺、智能發展遲緩,嬰幼兒時期食慾極差,因此生長發育均明顯落後,然而到6個月大至6歲之後,患兒的胃口逐漸改善,並發展出過度飲食的偏差現象,常會造成肥胖、糖尿病等併發症。
患兒必須自小即接受定期追蹤檢查,並視狀況給予特殊醫療及飲食指導,必要時尚需接受行為治療。患者通常有輕中度智障,也應接受早療及特殊教育,以提升其智能表現。
PWS患兒的肌肉張力差、皮膚及頭髮顏色較淺、智能發展遲緩,嬰幼兒時期食慾極差,因此生長發育均明顯落後,然而到6個月大至6歲之後,患兒的胃口逐漸改善,並發展出過度飲食的偏差現象,常會造成肥胖、糖尿病等併發症。
患兒必須自小即接受定期追蹤檢查,並視狀況給予特殊醫療及飲食指導,必要時尚需接受行為治療。患者通常有輕中度智障,也應接受早療及特殊教育,以提升其智能表現。
(2) 易脆X症候群
易脆X症候群(fragile X syndrome)又叫X脆折症,是最常見的性聯遺傳智能障礙疾病,大約每1,250位男性即可發現一例,女性的發生率較低,病情也較輕微。
實驗室檢查會顯現非常特殊的X染色體長臂尾端Xq27.3部位折斷的現象,因此得名。後來的研究發現,Xq27.3區段產生斷裂現象的原因與其中的一段三核 酸重複序列(CGG)n數目過度擴張有關,使得其鄰近的家族性智障基因(familial mental retardation-1; FMR-1 gene)受到干擾而致病。
正常人的(CGG)n數目在6~54之間,而無症狀帶因者則在54~200之譜,此狀態下的(CGG)n稱為先期突變(premutation),而當帶因者的兒女遺傳到有缺陷的X染色體時,(CGG)n會過度擴增到超過200,而進入病變狀態,便會抑制FMR-1基因的表現,造成種種的臨床症狀。
實驗室檢查會顯現非常特殊的X染色體長臂尾端Xq27.3部位折斷的現象,因此得名。後來的研究發現,Xq27.3區段產生斷裂現象的原因與其中的一段三核 酸重複序列(CGG)n數目過度擴張有關,使得其鄰近的家族性智障基因(familial mental retardation-1; FMR-1 gene)受到干擾而致病。
正常人的(CGG)n數目在6~54之間,而無症狀帶因者則在54~200之譜,此狀態下的(CGG)n稱為先期突變(premutation),而當帶因者的兒女遺傳到有缺陷的X染色體時,(CGG)n會過度擴增到超過200,而進入病變狀態,便會抑制FMR-1基因的表現,造成種種的臨床症狀。
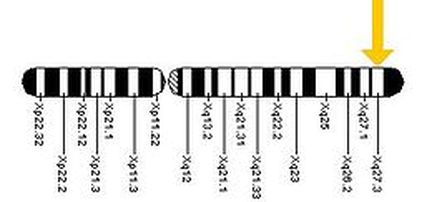
FMR-1基因所在的位置。
易脆X症有三大主徵,即智能障礙、臉長耳大及青春期後男性的巨睾症,其他尚有大鼻子、大下巴、前額突顯、關節過度伸展、僧帽瓣脫垂、過動及自閉傾向等。
此症無特殊治療方法,只能借助早期療育、特殊教育及支持性療法,及早安排適當的照顧計畫。家人應積極尋求遺傳諮詢的協助,以避免疾病的再發。
其他非染色體疾病,但也會因三核 酸重複序列過度擴張而導致的遺傳病尚有:因(CTG)n過度擴張(n≧50)所致的強直性肌肉失養症(myotonic dystrophy),因(CAG)n過度放大(n≧42)所致的亨丁頓氏舞蹈症(Huntington's chorea),以及因(CAG)n過度放大所致的第一、二、三型脊髓小腦萎縮症(spinocerebellar ataxia),會造成漸進性的運動協調障礙。
這一類遺傳性疾病均在青中年之後才發病,病徵發作時往往已有家庭及兒女,所引發的家庭及社會問題不容忽視,亟需醫學界、政府相關部門及社會資源的積極協助,方能解決無辜罹病者錯綜複雜的問題。
此症無特殊治療方法,只能借助早期療育、特殊教育及支持性療法,及早安排適當的照顧計畫。家人應積極尋求遺傳諮詢的協助,以避免疾病的再發。
其他非染色體疾病,但也會因三核 酸重複序列過度擴張而導致的遺傳病尚有:因(CTG)n過度擴張(n≧50)所致的強直性肌肉失養症(myotonic dystrophy),因(CAG)n過度放大(n≧42)所致的亨丁頓氏舞蹈症(Huntington's chorea),以及因(CAG)n過度放大所致的第一、二、三型脊髓小腦萎縮症(spinocerebellar ataxia),會造成漸進性的運動協調障礙。
這一類遺傳性疾病均在青中年之後才發病,病徵發作時往往已有家庭及兒女,所引發的家庭及社會問題不容忽視,亟需醫學界、政府相關部門及社會資源的積極協助,方能解決無辜罹病者錯綜複雜的問題。
對於患者,我們要抱持著正確的心態
遺傳醫學的進展,已能較有效的來協助病兒及家長找尋病因及預防再發,至於治療方式雖然多數尚處於症狀療育的階段,不過仍能緩解病情,或至少能讓不幸罹病者有尊嚴的活到生命的最後一刻。
家長必須學習接納與釋懷,社會大眾及醫療專業團隊也必須給予包容與尊重,協助病人及其家庭積極的面對事實,坦然接受疾病所帶來的各種挑戰,發揮人性的光明面。
家長必須學習接納與釋懷,社會大眾及醫療專業團隊也必須給予包容與尊重,協助病人及其家庭積極的面對事實,坦然接受疾病所帶來的各種挑戰,發揮人性的光明面。
